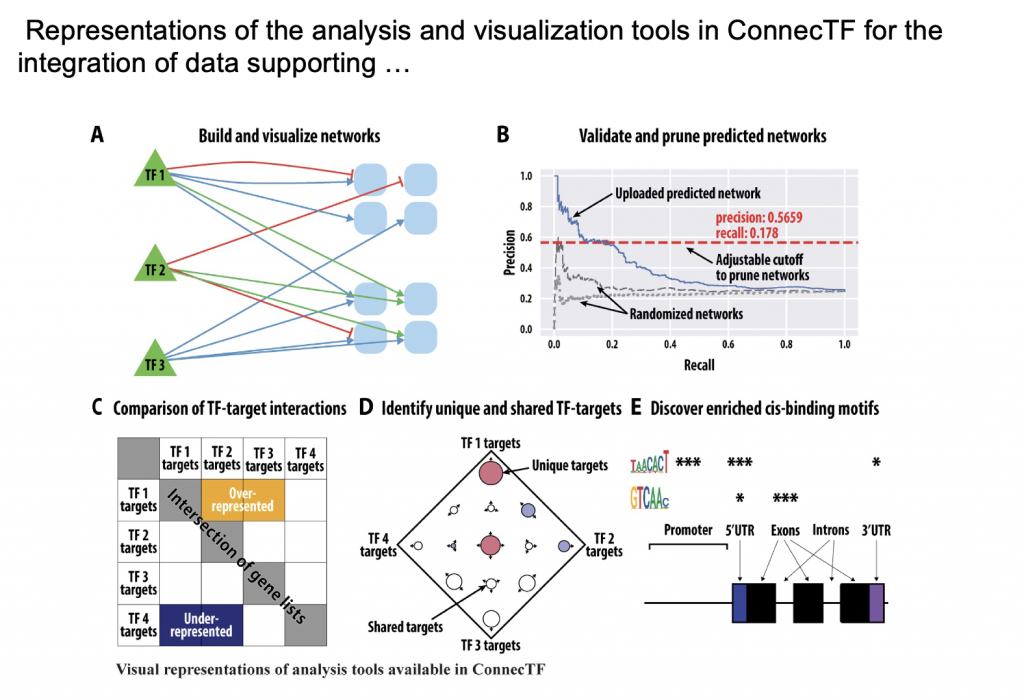
ConnecTF
Deciphering gene regulatory networks (GRNs) is both a promise and challenge of systems biology. The promise lies in identifying key transcription factors (TFs) that enable an organism to react to changes in its environment. The challenge lies in validating GRNs that involve hundreds of TFs with hundreds of thousands of interactions with their genome-wide targets experimentally determined by high-throughput sequencing. To address this challenge, we developed ConnecTF, a species-independent, web-based platform that integrates genome-wide studies of TF–target binding, TF–target regulation, and other TF-centric omic datasets and uses these to build and refine validated or inferred GRNs. We demonstrate the functionality of ConnecTF by showing how integration within and across TF–target datasets uncovers biological insights. Case study 1 uses integration of TF–target gene regulation and binding datasets to uncover TF mode-of-action and identify potential TF partners for 14 TFs in abscisic acid signaling. Case study 2 demonstrates how genome-wide TF–target data and automated functions in ConnecTF are used in precision/recall analysis and pruning of an inferred GRN for nitrogen signaling. Case study 3 uses ConnecTF to chart a network path from NLP7, a master TF in nitrogen signaling, to direct secondary TF2s and to its indirect targets in a Network Walking approach. The public version of ConnecTF (https://ConnecTF.org) contains 3,738,278 TF–target interactions for 423 TFs in Arabidopsis, 839,210 TF–target interactions for 139 TFs in maize (Zea mays), and 293,094 TF–target interactions for 26 TFs in rice (Oryza sativa). The database and tools in ConnecTF will advance the exploration of GRNs in plant systems biology applications for model and crop species.
Matthew D Brooks, Che-Lun Juang, Manpreet Singh Katari, José M Alvarez, Angelo Pasquino, Hung-Jui Shih, Ji Huang, Carly Shanks, Jacopo Cirrone, Gloria M Coruzzi, Plant Physiology, Volume 185, Issue 1, January 2021, Pages 49–66, https://doi.org/10.1093/plphys/kiaa012
VirtualPlant
Our long term goal is to understand how internal and external perturbations affect processes and networks controlling plant growth and development. In this project, we start with data integration of the known relationships among genes, proteins and molecules (extracted from public databases and/or generated with predictive algorithms) as well as experimental measurements under many different treatments. We go beyond data integration to conceptual integration by using novel visualization techniques to render the multivariate information in visual formats that facilitate extraction of biological concepts. We also use mathematical and statistical methods to help summarize the data. We implement and combine these approaches in a system we term “VirtualPlant”. Whereas our project relates specifically to Arabidopsis, the data structures, algorithms, and visualization tools are designed in a species-independent way. Thus the informatic, math, statistic and visualization tools that we develop can be used to model the cellular and physiological responses of any organism for which genomic data is available.
We have implemented a proto-type that is already being actively and effectively used (http://www.virtualplant.org). This tool is being used by biologists and computer scientist alike for the purpose it was designed for – to support the analysis of original genomic data generated by the researchers themselves. We have found that working with experimental biologists, even from very early stages of software development, to be the most effective way to generate real solutions to the problems encountered by researchers in the laboratory.
Biomaps: Find biological themes (Based on MIPS funcats or GO terms) in gene lists. This program analyses the distribution of functional assignments (Gene Ontology or MIPS) for one or more lists of genes. It reports back those terms that are found over-represented in the list(s) provided, as compared to the frequency of the term in a background population (e.g. the whole genome). A graphical and tabular output is given to facilitate analysis and interpretation of the results.
SunGear
Sungear is a generalized Venn Diagram. You can use this tool to compare an arbitrary number of lists or gene sets.
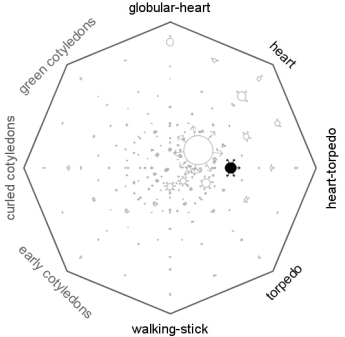 Sungear runs as an applet and can be started from the GeneCart. Sungear can be used to learn the biological significance of gene lists or intersections between gene lists. We have integrated Gene Names and Gene Ontology information to rapidly evaluate the significance of any intersection or selection made within Sungear. You can also hypothesize major trends in your data by using Sungear. The software can suggest biased GO terms (suggest over-representation) as well as identify the most “peculiar” intersections or gene sets based on the distribution of all GO terms associated with it.
Sungear runs as an applet and can be started from the GeneCart. Sungear can be used to learn the biological significance of gene lists or intersections between gene lists. We have integrated Gene Names and Gene Ontology information to rapidly evaluate the significance of any intersection or selection made within Sungear. You can also hypothesize major trends in your data by using Sungear. The software can suggest biased GO terms (suggest over-representation) as well as identify the most “peculiar” intersections or gene sets based on the distribution of all GO terms associated with it.
Please note: Sungear requires Java 1.4.2. You can download either the J2SE SDK (full install) or the J2SE JRE (runtime only), both of which can be found here. Be sure not to get the J2EE (Java 2 Enterprise Edition, as opposed to the Java 2 Standard Edition).
If the data set doesn’t load automatically:
Choose “File->Load->development” and click open to look at an example dataset generated from the AtGenExpress developmental time courses. This example shows genes enriched at the indicated developmental stages.
OrthologID
(OrthologID is undergoing updates and will be up soon)

This web-based tool is initiated by the Plant Genomics Consortium, and is designed to facilitate the identification of orthologous gene regions within a character-based phylogenetic framework. OrthologID will use a submitted sequence to query a local database to find all putative orthologs within the complete genomes of Oryza and Arabidopsis. Secondly, OrthologID will generate a gene tree (here referred to as a “guide tree”) of all putative orthologous gene regions from the complete genomes (as additional complete genomes become available, they will be added to the local database). It will assemble the matrix, remove redundant sequences, align sequences, perform tree searches using the parsimony ratchet and compute a strict consensus when multiple equally parsimonious trees are recovered. This guide tree will be probed for the presence of characters diagnostic of orthologous groups by passing the tree to the program P-Gnome (Sarkar et al., 2002). The submitted query sequence then will be screened for the presence of shared diagnostic characters using the program P-Elf (Sarkar et al., 2002). Finally, OrthologID will compare the output from P-Elf to the guide tree diagnostics and display the results as a tree with the query sequence appended to its ortholog.
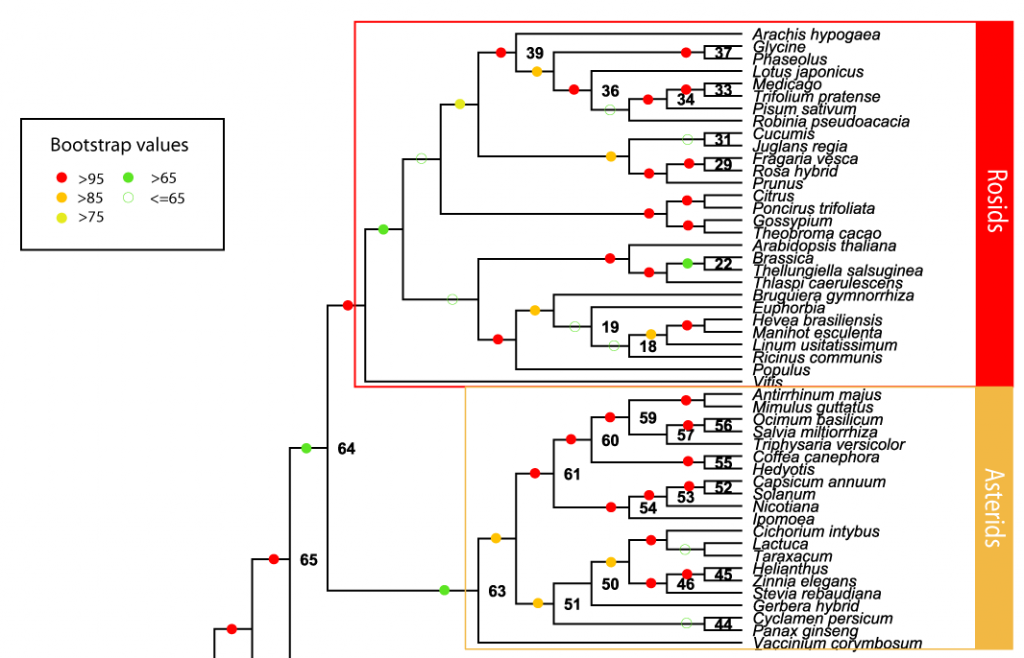
BigPlant
The BigPlant project uses a phylogenomic approach to produce a high resolution phylogeny of the seed plants. The current seed plant matrix comprises 150 species (including 5 outgroup taxa) and over 10 million amino acid characters from more than 22,000 genes. In its next iteration the BigPlant project will expand to include over 200 species with particular attention paid to filling in the less-represented clades in the current phylogeny. A collaborative effort across 4 premium institutes (http://nypg.bio.nyu.edu) is currently developing the molecular resources to fulfill this important goal.